Winrevair, para o que é indicado e para o que serve?
WinrevairTM é um medicamento sob prescrição para tratar adultos com hipertensão arterial pulmonar (HAP). A HAP é um tipo de pressão arterial elevada nas artérias dos pulmões.
WinrevairTM pode melhorar a sua capacidade de ser fisicamente ativo, reduzindo a pressão arterial em seus pulmões. Também pode aliviar os sintomas da HAP e retardar a progressão da sua doença.
Como o Winrevair funciona?
WinrevairTM é uma proteína de fusão do receptor de activina tipo IIA-Fc (ActRIIA-Fc) que atua reduzindo a pressão arterial nos pulmões.
Quais as contraindicações do Winrevair?
Não tome WinrevairTM se você é alérgico ao sotatercepte ou a qualquer outro componente deste medicamento.
Como usar o Winrevair?
Se o seu médico decidir que você ou um cuidador pode aplicar as injeções de WinrevairTM em casa, você ou seu cuidador deve receber treinamento de um profissional de saúde apropriado sobre a maneira correta de preparar e injetar WinrevairTM. Não tente injetar WinrevairTM até que um profissional de saúde lhe mostre a maneira correta.
O seu médico fará um exame de sangue antes das primeiras 5 doses de WinrevairTM, por mais tempo se necessário, e depois, de tempos em tempos, para verificar seus níveis de hemoglobina (uma proteína nos glóbulos vermelhos que transporta oxigênio) e plaquetas (células sanguíneas que ajudam o sangue a coagular). Após cada um desses exames de sangue, seu médico pode adiar o tratamento ou alterar sua dose, se necessário.
Leia o item "Instruções de Uso".
Use WinrevairTM exatamente como indicado pelo seu médico.
Você deverá usar WinrevairTM a cada 3 semanas como uma injeção logo abaixo da pele (subcutânea) apenas nestes locais de injeção:
Abdômen, com pelo menos 5 centímetros de distância do umbigo;
Parte superior da coxa;
Você deve aplicar a injeção de WinrevairTM imediatamente após misturar o medicamento em pó com a água para injetáveis, mas no máximo até 4 horas após a mistura.
Instruções de uso - kit com 1 frasco-ampola
Conhecendo os itens do seu kit
Informações importantes para saber antes da injeção
Você deve misturar este medicamento antes de utilizá-lo. Certifique-se de que o pó do medicamento no frasco esteja completamente dissolvido quando você injetar.
Verifique a dose prescrita (quantidade em “mL”) a cada vez que você usar o medicamento. A sua dose prescrita pode mudar.
Use somente os itens que acompanham o kit para preparar a dose prescrita.
Não abra a embalagem, nem misture o medicamento até estar pronto para utilizá-lo.
Não reutilize nenhum dos itens. Após aplicar a injeção, descarte o frasco usado com qualquer medicamento restante de WinrevairTM, agulha e seringas em um recipiente para objetos perfurocortantes.
Armazenando seu kit
Guarde todo o kit de injeção na geladeira (2°C a 8°C), mas não congele.
Mantenha o medicamento e os itens no kit de injeção e proteja-o da luz.
Mantenha o kit de injeção fora do alcance de crianças e animais de estimação.
Início
Qualquer paciente ou cuidador que preparar e injetar WinrevairTM deve primeiro ser treinado e considerado capaz por um profissional de saúde para administrar sozinho WinrevairTM.
1. Verificando o produto WinrevairTM e o prazo de validade
Retire o kit de injeção WinrevairTM da geladeira.
Verifique o prazo de validade e se a caixa ou os itens do kit apresentam algum dano.
Se vencido ou danificado, não use. Fale imediatamente com o seu médico ou farmacêutico.
Verifique se este é o medicamento prescrito pelo seu médico.
2. Aguardando seu kit atingir a temperatura ambiente, reunindo os itens e lavando suas mãos
Aguarde 15 minutos para permitir que o kit atinja a temperatura ambiente. Medicamento frio é mais dolorido ao injetar.
Junto com o seu kit, reúna esses itens e procure uma superfície limpa e plana para preparar e injetar a sua dose.
Lave suas mãos com água e sabão.
Misturando o medicamento em pó na forma líquida
Comece com a Bandeja Superior.
3. Removendo o frasco, a seringa preenchida e o adaptador do frasco do kit
4a. Verificando o frasco do medicamento
Não está danificado?
Sem partículas visíveis?
Está dentro da validade?
Verifique o rótulo do frasco para confirmar que o medicamento não está vencido.
Inspecione o pó do medicamento. Deve ser branco a esbranquiçado e pode parecer um pó compactado inteiro ou fragmentado.
Não use se estiver vencido, danificado ou se você puder ver partículas nele.
4b. Removendo a tampa plástica e limpando o frasco
Retire a tampa de plástico e limpe a tampa de borracha na parte superior do frasco com um lenço umedecido com álcool.
Não use se a tampa do frasco estiver faltando.
Não toque na tampa de borracha limpa.
Coloque o frasco em uma superfície limpa e plana.
5a. Encaixando o adaptador ao frasco
Abra a embalagem do adaptador do frasco e remova-o da embalagem.
Segure a parte azul do adaptador e encaixe o adaptador do frasco com a parte superior do frasco.
Não toque no interior do adaptador do frasco.
Para conectar o adaptador do frasco:
5b. Conectando o adaptador ao frasco
Segure o frasco com uma mão. Empurre para baixo com firmeza para que se encaixe no lugar (você pode sentir um pouco de resistência).
5c. Limpando o adaptador do frasco
Limpe a parte superior do adaptador do frasco com um lenço umedecido com álcool.
6. Verificando a seringa preenchida
Confirme se a água para injetáveis dentro da seringa preenchida está límpida e se o produto não está vencido.
Não danificada?
Límpida?
Sem partículas?
Está dentro da validade?
Não use se você observar grumos, partículas, alteração da coloração ou se o produto estiver vencido.
7. Retirando a tampa branca da seringa preenchida
Retire a tampa da seringa preenchida ao longo da perfuração.
Não toque na ponta da seringa preenchida.
Não empurre o êmbolo; pode derramar a água.
8. Conectando a seringa preenchida ao adaptador do frasco
Agora, pegue o frasco do medicamento com o adaptador do frasco acoplado.
Alinhe a ponta da seringa preenchida com o círculo azul do adaptador do frasco.
Empurre e gire a seringa preenchida no adaptador do frasco até não conseguir girar mais. Ao rosquear, certifique-se de segurar o adaptador do frasco.
Empurre e gire até conectar.
Não empurre o êmbolo; pode derramar a água.
9. Transferindo a água para injetáveis da seringa preenchida para o frasco
Empurre lentamente o êmbolo totalmente para baixo para transferir toda a água para injetáveis dentro do frasco (o êmbolo se moverá para cima; isso é normal).
10. Mexendo para misturar o medicamento
Não agite o frasco.
Segure a seringa preenchida e mexa suavemente o frasco em movimentos circulares até que o pó esteja completamente dissolvido. Isso pode levar até 2 minutos.
Quando o medicamento estiver bem misturado, deve ficar límpido. Caso contrário, repita esta etapa até que fique límpido.
Pressione o êmbolo para baixo para garantir que todo o líquido esteja no frasco (o êmbolo se moverá para cima; isso é normal).
11. Aguardando as bolhas desaparecerem
Deixe o frasco repousar para que as bolhas desapareçam. Isso pode levar até 3 minutos.
Antes de continuar, certifique-se de que o medicamento no frasco:
Está límpido a ligeiramente perolado e transparente a ligeiramente amarelo acastanhado.
Não tenha grumos ou pó.
Não tenha bolhas grandes.
Não há problema em ter uma leve espuma nas bordas do frasco.
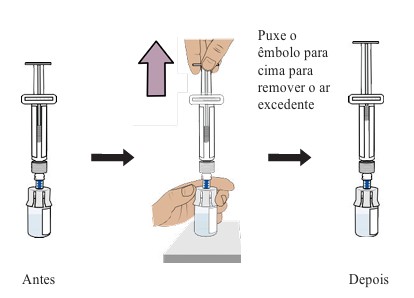
12. Preparando o frasco removendo o ar excedente
Enquanto o frasco estiver na posição vertical, puxe suavemente o êmbolo para cima até o topo do corpo, mas tenha cuidado para não puxar o êmbolo para fora da seringa.
Dica: Esta etapa apenas retira o ar excedente do frasco para garantir que você tenha a dose certa.
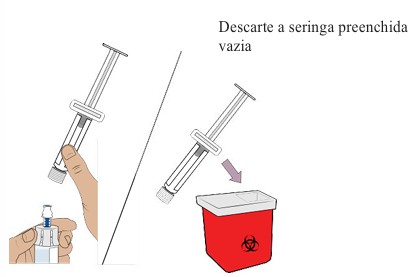
13. Removendo a seringa preenchida do frasco
Segure o adaptador do frasco e desrosqueie a seringa do frasco.
Descarte a seringa no recipiente para objetos perfurocortantes.
Você deve ter um frasco de medicamento preparado e pronto para ser usado nas próximas etapas.
Retirando a dose prescrita
Para as próximas etapas, você precisará de:
Frasco misturado de medicamento.
Itens da Bandeja Inferior.
14. Limpando a parte superior do adaptador do frasco
Com um novo lenço umedecido com álcool da bandeja inferior, limpe a parte superior do adaptador do frasco.
15. Removendo a seringa dosadora vazia da embalagem
Localize a seringa dosadora vazia na bandeja inferior e retire-a da embalagem.
Você usará esta seringa dosadora para medir o medicamento de que precisa (com base na dose prescrita).
16. Puxando o ar para a seringa dosadora
Você deve fazer isso para garantir que a pressão no frasco seja uniforme e obter uma dose precisa.
Segure a seringa dosadora na vertical e puxe o êmbolo para baixo para retirar o ar para dentro da seringa dosadora. Pare quando chegar à quantidade em “mL” descrita na sua prescrição.
17. Conectando a seringa dosadora ao frasco
Enquanto segura o adaptador do frasco, rosqueie a seringa dosadora até parar.
18. Empurrando o ar para dentro do frasco e virando de cabeça para baixo
Empurre o êmbolo totalmente para baixo para transferir todo o ar para o frasco.
Em seguida, segure o êmbolo no lugar com o dedo polegar e vire o frasco de cabeça para baixo.
19. Puxando o êmbolo de volta para retirar a sua dose
Com o frasco e a seringa dosadora virados para baixo, puxe lentamente o êmbolo de volta.
Pare quando chegar à quantidade em “mL” definida na sua prescrição.
20. Verificando se há bolhas de ar e bolsas de ar
Verifique se existem grandes bolhas de ar ou bolsas de ar na seringa. Você removerá o ar excedente nas próximas etapas.
21. Removendo bolhas de ar e bolsas de ar
Se você observar bolhas de ar ou uma bolsa de ar, bata na lateral da seringa dosadora para mover o ar para o topo.
Empurre lentamente o êmbolo para cima para remover o ar excedente.
22. Comparando a quantidade com a dose prescrita
Depois de remover todo o ar excedente, compare a quantidade com a dose prescrita.
Se não tiver a quantidade prescrita na seringa, puxe lentamente o êmbolo de volta para retirar mais medicamento.
Repita as etapas 19 a 21 até atingir a sua dose prescrita e não ter nenhuma bolha grande visível.
23. Confirmando a dose prescrita
Antes de continuar, verifique se você tem a dose prescrita na seringa dosadora.
Se a quantidade não corresponder à dose prescrita, repita as etapas 19 a 22.
24. Removendo a seringa dosadora do frasco e colocando-a ao lado
Segure o êmbolo no lugar com uma mão. Com a outra mão, segure o adaptador do frasco e desrosqueie a seringa dosadora preenchida do frasco.
Descarte o frasco no recipiente para objetos perfurocortantes.
Coloque a seringa dosadora preenchida sobre uma superfície plana e limpa.
Não toque na ponta da seringa dosadora, nem a deixe tocar em nenhuma superfície.
25. Conectando a agulha da injeção
Não toque no eixo de conexão da agulha.
Localize a agulha na bandeja inferior e abra a embalagem.
Com a agulha ainda na embalagem, segure a base da agulha e gire a seringa dosadora até parar. Retire a agulha da embalagem.
Afaste a proteção de segurança da agulha na direção da seringa no ângulo mostrado. Coloque a seringa dosadora sobre uma superfície plana e limpa.
Não destampe a agulha.
26. Escolhendo e limpando o local de aplicação da injeção
Selecione um local de aplicação da injeção na barriga (abdômen) ou na parte superior da coxa. Se injetar na área do abdômen, aplique a injeção com uma distância de 5 cm ao redor do seu umbigo.
Escolha um local diferente cada vez que injetar.
Não realize a injeção na pele ferida, dolorida, machucada ou com manchas vermelhas;
Não injete através da roupa.
Limpe o local da injeção com um novo lenço umedecido com álcool.
Não toque novamente no local limpo onde será aplicada a injeção.
Agora você está pronto para injetar o medicamento.
27. Injetando o medicamento
Puxe a tampa diretamente para fora da agulha.
Descarte a tampa.
Não toque no êmbolo até estar pronto para injetar, para não perder nenhum medicamento.
Aperte suavemente e segure uma dobra da pele onde você irá injetar. Insira a agulha com um movimento semelhante a um dardo em um ângulo de 45° a 90°. Isto ajuda a injetar diretamente sob a pele (injeção subcutânea).
Empurre o êmbolo com pressão lenta e constante até que a seringa dosadora esteja vazia.
Confirme que todo o medicamento foi injetado.
Agora você pode soltar a dobra da pele.
Sempre mantenha os dedos afastados da agulha.
Mantendo o êmbolo pressionado, remova a agulha da pele no mesmo ângulo em que a inseriu.
Para reaplicar a proteção de segurança da agulha, empurre-a contra uma superfície plana até ouvir um “clique” e ver que a agulha está coberta.
Descarte a seringa dosadora e os itens usados em um recipiente para descarte de objetos perfurocortantes.
Não remova a agulha da seringa dosadora.
Como descartar WinrevairTM
Descarte qualquer frasco usado (incluindo qualquer líquido restante de WinrevairTM), agulha, tampas de frasco e de agulha e seringas usadas em um recipiente para descarte de objetos perfurocortantes.
Não descarte os frascos, seringas ou agulhas de WinrevairTM no lixo doméstico.
Não reutilize nenhum dos itens. Este produto é descartável e deve ser usado apenas uma vez.
Importante: Mantenha sempre o recipiente para descarte de objetos perfurocortantes fora do alcance de crianças e animais de estimação.
Se você não tiver um recipiente para descarte de objetos perfurocortantes, poderá usar um recipiente doméstico que seja:
Feito de um plástico resistente;
Que pode ser fechado com uma tampa hermética e resistente a perfurações, sem que objetos pontiagudos possam sair;
Firme e estável durante o uso;
Resistente a vazamentos; e;
Devidamente rotulado para alertar sobre resíduos perigosos dentro do recipiente.
Quando o recipiente para descarte de objetos perfurocortantes estiver quase cheio, você precisará seguir as diretrizes locais sobre a maneira correta de descartar o recipiente de objetos perfurocortantes. Não jogue fora medicamentos, frascos, agulhas soltas ou seringas no lixo doméstico. Pergunte ao seu farmacêutico como jogar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
Não recicle o seu recipiente usado de objetos perfurocortantes.
Instruções de uso - kit com 2 frascos-ampola
Conhecendo os itens de seu kit
Informações importantes para saber antes da injeção
Você deve misturar este medicamento antes de utilizá-lo. Certifique-se de que o pó do medicamento no frasco esteja completamente dissolvido quando você injetar.
Verifique a dose prescrita (quantidade em “mL”) a cada vez que você usar o medicamento. A sua dose prescrita pode mudar.
Use somente os itens que acompanham o kit para preparar a dose prescrita.
Não abra a embalagem, nem misture o medicamento até estar pronto para utilizá-lo.
Não reutilize nenhum dos itens. Após aplicar a injeção, descarte o frasco usado com qualquer medicamento restante de WinrevairTM, agulha e seringas em um recipiente para objetos perfurocortantes.
Armazenando seu kit
Guarde todo o kit de injeção na geladeira (2°C a 8°C), mas não congele.
Mantenha o medicamento e os itens no kit de injeção e proteja-o da luz.
Mantenha o kit de injeção fora do alcance de crianças e animais de estimação.
Início
Qualquer paciente ou cuidador que preparar e injetar WinrevairTM deve primeiro ser treinado e considerado capaz por um profissional de saúde para administrar sozinho WinrevairTM.
1. Verificando o produto WinrevairTM e o prazo de validade
Retire o kit de injeção WinrevairTM da geladeira.
Verifique o prazo de validade e se a caixa ou os itens do kit apresentam algum dano.
Se vencido ou danificado, não use. Fale imediatamente com o seu médico ou farmacêutico.
Verifique se este é o medicamento prescrito pelo seu médico.
2. Aguardando seu kit atingir a temperatura ambiente, reunindo os itens e lavando suas mãos
Aguarde 15 minutos para permitir que o kit atinja a temperatura ambiente. Medicamento frio é mais dolorido ao injetar.
Junto com o seu kit, reúna esses itens e procure uma superfície limpa e plana para preparar e injetar a sua dose.
Lave suas mãos com água e sabão.
Misturando o medicamento em pó na forma líquida
Comece com a Bandeja Superior.
3. Removendo o frasco 1, a seringa preenchida 1 e o adaptador do frasco 1 do kit
4a. Verificando o frasco do medicamento
Não está danificado?
Sem partículas visíveis?
Está dentro da validade?
Verifique o rótulo do frasco para confirmar que o medicamento não está vencido.
Inspecione o pó do medicamento. Deve ser branco a esbranquiçado e pode parecer um pó compactado inteiro ou fragmentado.
Não use se estiver vencido, danificado ou se você puder ver partículas nele.
4b. Removendo a tampa plástica e limpando o frasco
Retire a tampa de plástico e limpe a tampa de borracha na parte superior do frasco com um lenço umedecido com álcool.
Não use se a tampa do frasco estiver faltando.
Não toque na tampa de borracha limpa.
Coloque o frasco em uma superfície limpa e plana.
5a. Encaixando o adaptador ao frasco
Abra a embalagem do adaptador do frasco e remova-o da embalagem.
Segure a parte azul do adaptador e encaixe o adaptador do frasco com a parte superior do frasco.
Não toque no interior do adaptador do frasco.
Para conectar o adaptador do frasco:
5b. Conectando o adaptador ao frasco
Segure o frasco com uma mão. Empurre para baixo com firmeza para que se encaixe no lugar (você pode sentir um pouco de resistência).
5c. Limpando o adaptador do frasco
Limpe a parte superior do adaptador do frasco com um lenço umedecido com álcool.
6. Verificando a seringa preenchida
Confirme se a água para injetáveis dentro da seringa preenchida está límpida e se o produto não está vencido.
Não danificada?
Límpida?
Sem partículas?
Está dentro da validade?
Não use se você observar grumos, partículas, alteração da coloração ou se o produto estiver vencido.
7. Retirando a tampa branca da seringa preenchida
Retire a tampa da seringa preenchida ao longo da perfuração.
Não toque na ponta da seringa preenchida.
Não empurre o êmbolo; pode derramar a água.
8. Conectando a seringa preenchida ao adaptador do frasco
Agora, pegue o frasco do medicamento com o adaptador do frasco acoplado.
Alinhe a ponta da seringa preenchida com o círculo azul do adaptador do frasco.
Empurre e gire a seringa preenchida no adaptador do frasco até não conseguir girar mais. Ao rosquear, certifique-se de segurar o adaptador do frasco.
Empurre e gire até conectar.
Não empurre o êmbolo; pode derramar a água.
9. Transferindo a água para injetáveis da seringa preenchida para o frasco
Empurre lentamente o êmbolo totalmente para baixo para transferir toda a água para injetáveis dentro do frasco (o êmbolo se moverá para cima; isso é normal).
Deixe a seringa preenchida acoplada ao frasco e coloque sobre uma superfície plana.
10. Preparando o frasco 2, a seringa preenchida 2 e o adaptador do frasco 2
Remova o frasco 2, a seringa preenchida 2 e o adaptador do frasco 2 do kit.
Importante: Repita as etapas 4 a 9 para preparar o frasco 2.
Pare e verifique se você tem o frasco 1 e o frasco 2 preparados.
Você precisa de dos dois frascos preparados antes de poder continuar para a Etapa 11.
11. Mexendo para misturar o medicamento
Não agite os frascos
Segure as seringas preenchidas em cada mão e mexa suavemente os frascos em movimentos circulares até que o pó esteja completamente dissolvido. Isso pode levar até 2 minutos.
Quando o medicamento estiver bem misturado, deve ficar límpido. Caso contrário, repita esta etapa até que fique límpido.
Para cada frasco, pressione o êmbolo para baixo para garantir que todo o líquido esteja no frasco (o êmbolo se moverá para cima; isso é normal).
12. Aguardando as bolhas desaparecerem
Deixe o frasco repousar para que as bolhas desapareçam.
Isso pode levar até 3 minutos.
Antes de continuar, certifique-se de que o medicamento no frasco:
Está límpido a ligeiramente perolado e transparente a ligeiramente amarelo acastanhado;
Não tenha grumos ou pó;
Não tenha bolhas grandes.
Não há problema em ter uma leve espuma nas bordas do frasco.
13. Preparando os frascos removendo o ar excedente
Comece com qualquer frasco. Enquanto o frasco estiver na posição vertical, puxe suavemente o êmbolo para cima até o topo do corpo, mas tenha cuidado para não puxar o êmbolo para fora da seringa.
Importante: Execute esta etapa para ambos os frascos. Pare e verifique se ambos os frascos estão preparados antes de continuar
14. Removendo as seringas preenchidas dos frascos
Segure os adaptadores dos frascos e desrosqueie ambas as seringas dos frascos.
Descarte ambas as seringas no recipiente para objetos perfurocortantes.
Você deve ter 2 frascos de medicamentos preparados e prontos para serem combinados nas próximas etapas.
Combinando o medicamento dos dois frascos
Para as próximas etapas, você precisará de:
Frascos 1 e 2 misturados;
Itens da Bandeja Inferior.
15. Limpando a parte superior de ambos os adaptadores do frasco
Com 2 lenços umedecidos com álcool da bandeja inferior, limpe a parte superior dos adaptadores dos frascos.
16. Removendo a seringa dosadora vazia da embalagem
Localize a seringa dosadora vazia na bandeja inferior e retire-a da embalagem.
Você usará esta seringa dosadora para medir o medicamento de que você precisa (com base na dose prescrita).
17. Puxando o ar para a seringa dosadora
Você deve fazer isso para garantir que a pressão no frasco seja uniforme e obter uma dose precisa.
Segure a seringa dosadora na vertical e puxe o êmbolo para baixo para aspirar 1,5 mL de ar.
18. Conectando a seringa dosadora em um dos frascos
Enquanto segura o adaptador do frasco, rosqueie a seringa dosadora até parar.
19. Empurrando o ar para dentro do frasco e virando de cabeça para baixo
Empurre o êmbolo totalmente para baixo para transferir todo o ar para o frasco.
Em seguida, segure o êmbolo no lugar com o dedo polegar e vire o frasco de cabeça para baixo.
20. Retirando todo o medicamento do 1° frasco
Puxe lentamente o êmbolo para trás. Pare em 1,5 mL.
Isto garantirá que todo o medicamento do frasco estará na seringa dosadora.
Não puxe ar excedente para dentro da seringa. Pare em 1,5 mL.
Tenha cuidado para não puxar o êmbolo da seringa dosadora para fora.
21. Removendo o 1° frasco da seringa dosadora
Segure o adaptador do frasco e desrosqueie a seringa dosadora cheia do frasco.
Descarte o frasco vazio no recipiente para objetos perfurocortantes.
22. Conectando a seringa dosadora ao 2° frasco
Enquanto você segura o adaptador do frasco para injetáveis do 2º frasco, rosqueie a seringa dosadora parcialmente cheia no adaptador do frasco para injetáveis até parar.
23. Colocando todo o medicamento no 2° frasco e virando de cabeça para baixo
Empurre lentamente e totalmente o êmbolo para baixo, para transferir todo o medicamento para o frasco. Isto combina o medicamento de ambos os frascos.
Segure o êmbolo no lugar com o dedo polegar e vire o frasco de cabeça para baixo.
24. Puxando o êmbolo de volta para retirar a sua dose
Com o frasco e a seringa dosadora virados de cabeça para baixo, puxe lentamente o êmbolo de volta.
Pare quando chegar à quantidade em “mL” definida na sua prescrição.
25. Verificando se há bolhas de ar e bolsas de ar
Verifique se existem grandes bolhas de ar ou bolsas de ar na seringa. Você removerá o ar excedente nas próximas etapas.
26. Removendo bolhas de ar e bolsas de ar
Se você observar bolhas de ar ou uma bolsa de ar, bata na lateral da seringa dosadora para mover o ar para o topo.
Empurre lentamente o êmbolo para cima para remover o ar excedente.
27. Comparando a quantidade com a dose prescrita
Depois de remover todo o ar excedente, compare a quantidade com a dose prescrita.
Se não tiver a quantidade prescrita na seringa, puxe lentamente o êmbolo de volta para retirar mais medicamento.
Repita as etapas 24 a 26 até atingir a sua dose prescrita e não ter nenhuma bolha grande visível.
28. Confirmando a dose prescrita
Antes de continuar, verifique se você tem a dose prescrita na seringa dosadora.
Se a quantidade não corresponder à dose prescrita, repita as etapas 24 a 27.
29. Removendo a seringa dosadora do frasco e colocando-a ao lado
Segure o êmbolo no lugar com uma mão. Com a outra mão, segure o adaptador do frasco e desrosqueie a seringa dosadora preenchida do frasco.
Descarte o frasco no recipiente para objetos perfurocortantes.
Coloque a seringa dosadora preenchida sobre uma superfície plana e limpa.
Não toque na ponta da seringa dosadora, nem a deixe tocar em nenhuma superfície.
30. Conectando a agulha da injeção
Não toque no eixo de conexão da agulha.
Localize a agulha na bandeja inferior e abra a embalagem.
Com a agulha ainda na embalagem, segure a base da agulha e gire a seringa dosadora até parar. Retire a agulha da embalagem.
Afaste a proteção de segurança da agulha na direção da seringa no ângulo mostrado. Coloque a seringa dosadora sobre uma superfície plana e limpa.
Não destampe a agulha.
31. Escolhendo e limpando o local de aplicação da injeção
Selecione um local de aplicação da injeção na barriga (abdômen) ou na parte superior da coxa. Se injetar na área do abdômen, aplique a injeção com uma distância de 5 cm ao redor do seu umbigo.
Escolha um local diferente cada vez que injetar.
Não realize a injeção na pele ferida, dolorida, machucada ou com manchas vermelhas;
Não injete através da roupa.
Limpe o local da injeção com um novo lenço umedecido com álcool.
Não toque novamente no local limpo onde será aplicada a injeção.
Agora você está pronto para injetar o medicamento.
32. Injetando o medicamento
Puxe a tampa diretamente para fora da agulha.
Descarte a tampa.
Não toque no êmbolo até estar pronto para injetar, para não perder nenhum medicamento.
Aperte suavemente e segure uma dobra da pele onde você irá injetar. Insira a agulha com um movimento semelhante a um dardo em um ângulo de 45° a 90°. Isto ajuda a injetar diretamente sob a pele (injeção subcutânea).
Empurre o êmbolo com pressão lenta e constante até que a seringa dosadora esteja vazia.
Confirme que todo o medicamento foi injetado.
Agora você pode soltar a dobra da pele.
Sempre mantenha os dedos afastados da agulha.
Mantendo o êmbolo pressionado, remova a agulha da pele no mesmo ângulo em que a inseriu.
Para reaplicar a proteção de segurança da agulha, empurre-a contra uma superfície plana até ouvir um “clique” e ver que a agulha está coberta.
Descarte a seringa dosadora e os itens usados em um recipiente para descarte de objetos perfurocortantes.
Não remova a agulha da seringa dosadora.
Como descartar WinrevairTM
Descarte qualquer frasco usado (incluindo qualquer líquido restante de WinrevairTM), agulha, tampas de frasco e de agulha e seringas usadas em um recipiente para descarte de objetos perfurocortantes.
Não descarte os frascos, seringas ou agulhas de WinrevairTM no lixo doméstico.
Não reutilize nenhum dos itens. Este produto é descartável e deve ser usado apenas uma vez.
Importante: Mantenha sempre o recipiente para descarte de objetos perfurocortantes fora do alcance de crianças e animais de estimação.
Se você não tiver um recipiente para descarte de objetos perfurocortantes, poderá usar um recipiente doméstico que seja:
Feito de um plástico resistente;
Que pode ser fechado com uma tampa hermética e resistente a perfurações, sem que objetos pontiagudos possam sair;
Firme e estável durante o uso;
Resistente a vazamentos; e;
Devidamente rotulado para alertar sobre resíduos perigosos dentro do recipiente.
Quando o recipiente para descarte de objetos perfurocortantes estiver quase cheio, você precisará seguir as diretrizes locais sobre a maneira correta de descartar o recipiente de objetos perfurocortantes. Não jogue fora medicamentos, frascos, agulhas soltas ou seringas no lixo doméstico. Pergunte ao seu farmacêutico como jogar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
Não recicle o seu recipiente usado de objetos perfurocortantes.
Perguntas frequentes
O que devo fazer se estiver sangrando no local da injeção?
Coloque um algodão ou curativo na pele imediatamente e aplique uma pequena pressão. Se o sangramento não parar, ligue para o seu médico ou farmacêutico imediatamente.
Onde posso encontrar a quantidade prescrita a ser injetada?
A quantidade de prescrição em “mL” está na sua prescrição. Contate seu médico ou farmacêutico se você não conseguir encontrar a quantidade a ser injetada.
O que devo fazer se acidentalmente algum medicamento entrar em contato com a minha pele ou superfície do corpo?
Lave bem a área com água e sabão imediatamente.
O que devo fazer se não tiver certeza de que administrei corretamente a dose prescrita?
Ligue para o seu médico ou farmacêutico.
O que devo fazer se o êmbolo da minha seringa dosadora se mover automaticamente quando tento retirar o medicamento do frasco?
Não se preocupe se o êmbolo se mover ligeiramente sozinho quando você estiver enchendo a seringa dosadora com o medicamento.
Com uma mão, segure o êmbolo no lugar para impedir que ele se mova.
Com a outra mão, desrosqueie o frasco -ampola da seringa dosadora. Depois de desrosqueado, é seguro soltar o êmbolo.
Você pode evitar esse movimento automático do êmbolo empurrando ar para dentro do frasco antes de encher a seringa dosadora com o medicamento. Consulte as etapas 16 a 23 (kit com 1 frasco-ampola) ou 17 a 28 (kit com dois frascos-ampola) para obter instruções detalhadas.
O que devo fazer se os itens do meu kit estiverem danificados ou comprometidos (por exemplo, descoloridos, turvos ou com partículas)?
Se os itens do seu kit estiverem danificados ou comprometidos, não o use. Ligue para o seu médico ou farmacêutico.
O que devo fazer se o meu medicamento não ficar límpido depois de misturar e fazer movimentos circulares?
Não use o medicamento se você fez movimentos circulares no frasco do medicamento por cerca de 2 minutos e deixou-o repousar por mais 3 minutos, mas o frasco do medicamento permanece turvo ou apresenta grumos, pó ou partículas estranhas. Ligue para o seu médico ou farmacêutico.
O que devo fazer se a água para injetáveis não sair da seringa preenchida?
Verifique se o adaptador do frasco está firmemente conectado ao frasco. Caso contrário, segure o frasco e pressione firmemente o adaptador do frasco para garantir que o adaptador do frasco perfure a tampa de borracha do frasco.
O que devo fazer se eu deixar cair os componentes do meu kit?
Não use se algum item estiver danificado. Se não tiver certeza, ligue para seu médico ou farmacêutico.
Posso usar meu kit que ficou fora da geladeira?
Se o kit de injeção não utilizado estiver fora da geladeira por um longo período de tempo, entre em contato com seu médico ou farmacêutico antes de continuar.
Preciso usar o medicamento misturado imediatamente?
Recomendamos que você injete o medicamento logo após a mistura, mas não mais de 4 horas depois de misturado. Se passarem mais de 4 horas, descarte o medicamento. Se você tiver dúvidas ou não tiver certeza sobre o processo, entre em contato com seu médico ou farmacêutico.
Como posso obter ajuda para preparar e administrar minha injeção?
Se você tiver dúvidas sobre como administrar WinrevairTM da maneira correta ou precisar de mais informações, ligue para seu médico ou farmacêutico.
Para qualquer outra informação sobre este medicamento, contate o seu médico ou farmacêutico ou o serviço de atendimento do consumidor da empresa através do número 0800-0122232.
A sua dose:
A sua dose de WinrevairTM depende do seu peso corporal e exames de sangue.
O seu médico lhe dirá quanto de WINREVAIRTM deve aplicar e quando aplicá-lo.
Não altere a sua dose ou pare de aplicar WINREVAIRTM sem falar com o seu médico.
Não aplique WINREVAIRTM com mais frequência do que o indicado pelo seu médico. Se você não tiver certeza de quando aplicar WINREVAIRTM, consulte seu médico.
O seu médico monitorará a sua dose:
Antes da sua primeira dose e regularmente enquanto estiver usando WINREVAIRTM, o seu médico solicitará exames de sangue. Estes são feitos para que o seu médico possa te monitorar e encontrar a melhor dose para você.
O seu médico pode ajustar a sua dose, atrasar o tratamento ou interromper o tratamento, dependendo da sua resposta à WINREVAIRTM.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
O que devo fazer quando me esquecer de usar o Winrevair?
Se você perder a dose prescrita de WinrevairTM, aplique-a dentro de 3 dias e siga o cronograma original para a próxima dose. Se não for aplicada dentro de 3 dias, converse com seu médico para orientação.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Quais cuidados devo ter ao usar o Winrevair?
O que devo dizer ao meu médico antes e durante a utilização de WinrevairTM?
Informe ao seu médico (ou farmacêutico) sobre quaisquer condições médicas que você tem ou teve, e sobre quaisquer alergias.
Gravidez
WinrevairTM pode prejudicar o feto. Informe ao seu médico se estiver gestante ou se planeja engravidar.
Informe ao seu médico imediatamente se engravidar ou se pensar que pode estar gestante enquanto estiver utilizando WinrevairTM.
O seu médico deve solicitar um teste de gravidez antes de começar a utilizar WinrevairTM.
Você deve usar método contraceptivo eficaz ao utilizar WinrevairTM.
Você deve continuar a utilizar métodos contraceptivos eficazes durante, pelo menos, 4 meses após a sua última dose se você parar de tomar WinrevairTM. Pergunte ao seu médico sobre métodos contraceptivos que funcionariam bem para você.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Amamentação
Informe ao seu médico se estiver amamentando ou se planeja amamentar. Não se sabe se WinrevairTM passa para o leite materno.
Não amamentar enquanto estiver tomando WinrevairTM.
Não amamente durante pelo menos 4 meses após a sua última dose se você parar de tomar WinrevairTM. Converse com seu médico sobre a melhor maneira de alimentar o seu bebê.
Uso criterioso no aleitamento ou na doação de leite humano: O uso deste medicamento no período da lactação depende da avaliação e acompanhamento do seu médico ou cirurgião-dentista.
Crianças
Não se sabe se WinrevairTM é seguro e eficaz em crianças menores de 18 anos de idade.
Este medicamento pode causar doping.
Quais as reações adversas e os efeitos colaterais do Winrevair?
Qualquer medicamento pode ter efeitos não intencionais ou indesejáveis, os chamados efeitos colaterais.
WinrevairTM pode causar efeitos colaterais graves, incluindo:
Alto nível de hemoglobina (uma proteína nos glóbulos vermelhos que transporta oxigênio). Isso pode aumentar a chance de formação de um coágulo sanguíneo que pode bloquear um vaso sanguíneo. O seu médico solicitará exames de sangue para verificar os níveis de hemoglobina antes de começar e regularmente durante o tratamento com WinrevairTM.
Baixo número de plaquetas (células sanguíneas que ajudam a coagular o sangue). Isso pode causar sintomas como aparecimento fácil de manchas roxas, sangramento contínuo por cortes e sangramentos nasais. O seu médico solicitará exames de sangue para verificar o nível de plaquetas antes de começar e regularmente durante o tratamento com WinrevairTM. Informe o seu profissional de saúde se você desenvolver hematomas ou sangramentos facilmente, sangramento contínuo ou sangramento nasal.
Sangramento grave. Informe o seu profissional de saúde se desenvolver quaisquer sinais ou sintomas de sangramento, incluindo: vômito com sangue ou se seu vômito parecer borra de café; náusea; tontura ou sensação de fraqueza; urina rosa ou marrom; cólicas abdominais persistentes; fezes vermelhas ou pretas que parecem alcatrão; forte dor nas costas; tossir sangue ou coágulos sanguíneos; sangramento menstrual anormalmente intenso; dores de cabeça persistentes.
Diminuição da fertilidade. WinrevairTM pode diminuir a fertilidade feminina e masculina.
Os efeitos colaterais mais comuns de WinrevairTM são:
Dor de cabeça;
Sangramentos nasais (epistaxe);
Telangiectasia (também chamados de vasinhos ou pequenos vasos sanguíneos que se parecem com linhas rosas ou vermelhas na pele);
Diarreia;
Tontura;
Irritação na pele;
Aumento da pressão arterial;
Vermelhidão.
Outros efeitos colaterais também podem ocorrer raramente, e como com qualquer medicamento sob prescrição, alguns efeitos colaterais podem ser graves.
Pergunte ao seu médico ou farmacêutico para obter mais informações. Ambos têm uma lista mais completa de efeitos colaterais. Informe imediatamente ao seu médico ou farmacêutico sobre estes ou quaisquer outros sintomas incomuns.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, informe seu médico ou cirurgião-dentista.
Apresentações do Winrevair
Pó liofilizado para solução injetável 45mg e 60mg
45 mg de sotatercepte em frasco-ampola de dose única em embalagem com 1 frasco-ampola, 1 seringa preenchida de diluente, 1 adaptador, 1 seringa, 1 agulha e 4 envelopes com lenços umedecidos com álcool.
45 mg de sotatercepte em frasco-ampola de dose única em embalagem com 2 frascos-ampolas, 2 seringas preenchidas de diluente, 2 adaptadores, 1 seringa, 1 agulha e 8 envelopes com lenços umedecidos com álcool.
60 mg de sotatercepte em frasco-ampola de dose única em embalagem com 1 frasco-ampola, 1 seringa preenchida de diluente, 1 adaptador, 1 seringa, 1 agulha e 4 envelopes com lenços umedecidos com álcool.
60 mg de sotatercepte em frasco-ampola de dose única em embalagem com 2 frascos-ampolas, 2 seringas preenchidas de diluente, 2 adaptadores, 1 seringa, 1 agulha e 8 envelopes com lenços umedecidos com álcool.
Uso subcutâneo.
Uso adulto.
Qual a composição do Winrevair?
Frasco-ampola de dose única de 45 mg
Cada frasco contém 45 mg de sotatercepte. Após reconstituição com 1,0 mL de Água para Injetáveis, a concentração resultante é de 50 mg/1,0 mL de sotatercepte e o volume nominal de entrega é de 0,9 mL.
Cada frasco contém 60 mg de sotatercepte. Após reconstituição com 1,3 mL de Água para Injetáveis, a concentração resultante é de 50 mg/1,0 mL de sotatercepte e o volume nominal de entrega é de 1,2 mL.
Superdose: o que acontece se tomar uma dose do Winrevair maior do que a recomendada?
Se usar WinrevairTM em excesso, contate o seu médico ou farmacêutico.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação medicamentosa: quais os efeitos de tomar Winrevair com outros remédios?
Informe ao seu médico sobre todos os medicamentos que você toma, incluindo medicamentos sob prescrição e os isentos de prescrição, vitaminas e fitoterápicos.
Conheça os medicamentos que você toma. Mantenha uma lista deles e mostre a lista ao seu médico e farmacêutico quando receber um novo medicamento.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Qual a ação da substância do Winrevair?
Resultados de Eficácia
Hipertensão arterial pulmonar em indivíduos adultos
A eficácia de Sotatercepte foi avaliada em pacientes adultos com HAP no estudo STELLAR. O STELLAR foi um estudo clínico global, duplo-cego, controlado por placebo, multicêntrico, de grupos paralelos, no qual 323 pacientes com HAP (Grupo 1 CF II ou III da OMS) foram randomizados 1:1 para receber Sotatercepte (dose alvo de 0,7 mg/kg) (n=163) ou placebo (n=160) administrado por via subcutânea uma vez a cada 3 semanas.
As características demográficas e clínicas iniciais foram geralmente comparáveis entre os grupos Sotatercepte e placebo. Os participantes deste estudo eram adultos com idade mediana de 48,0 anos (intervalo: de 18 a 82 anos); peso médio 68 kg (intervalo: de 38,0 a 141,3 kg); 89,2% dos participantes eram brancos e 79,3% não eram hispânicos ou latinos; e 79,3% eram do sexo feminino. As etiologias mais comuns de HAP foram HAP idiopática (58,5%), HAP hereditária (18,3%) e HAP associada a doenças do tecido conjuntivo (DTC) (14,9%). O tempo médio entre o diagnóstico de HAP e a seleção foi de 8,76 anos. A maioria dos participantes estava recebendo terapia de base tripla (61,3%) ou dupla (34,7%) para HAP, e mais de um terço (39,9%) estava recebendo infusões de prostaciclina. Todos os pacientes estavam recebendo terapia de base estável para HAP há pelo menos 90 dias e continuaram assim durante todo o estudo. As proporções de participantes em CF II da OMS (48,6%) e em CF III da OMS (51,4%) foram semelhantes em ambos os grupos.
O estudo STELLAR excluiu pacientes com diagnóstico de hipertensão pulmonar (HP) dos Grupos 2, 3, 4 ou 5 da OMS; HAP associada ao vírus da imunodeficiência humana (HIV), HAP associada à hipertensão portal, HAP associada à esquistossomose e doença veno-oclusiva pulmonar. Outros critérios de exclusão incluíram, mas não se limitaram a: nível de hemoglobina acima do limite superior de normalidade específico por gênero na triagem, conforme teste laboratorial local; contagem basal de plaquetas <50.000/mm3 (<50,0 × 109/L) na triagem; hipertensão sistêmica não controlada, evidenciada por pressão arterial (PA) sistólica sentada >160 mmHg ou PA diastólica sentada >100 mmHg durante a visita após um período de repouso; PA sistólica basal <90 mmHg na triagem.
O desfecho primário de eficácia foi a mudança em relação ao valor basal na Semana 24 na distância no teste de caminhada de 6 minutos (TC6M). No grupo de tratamento Sotatercepte, a mediana da mudança ajustada por placebo no TC6M em relação ao valor basal na Semana 24, foi de 40,8 metros (IC 95%: 27,5; 54,1; p < 0,001). A mediana das mudanças ajustadas por placebo no TC6M na Semana 24, também foi avaliada em subgrupos (veja Figura 1).
Figura 1: Mudança na distância do teste de caminhada de 6 minutos (metros) basal na Semana 24 em subgrupos
A mudança em relação à linha de base no TC6M na Semana 24 para os indivíduos que morreram recebeu um valor de -2000 metros para receber a pior classificação. A mudança em relação à linha de base no TC6M na Semana 24 para indivíduos que faltaram dados devido a um evento de piora clínica não fatal foi imputada a -1000 metros para receber a próxima pior classificação.
A melhora clínica foi um desfecho pré-definido medido pela proporção de pacientes que atingiram todos os três seguintes critérios na Semana 24 em relação ao valor basal: melhora no TC6M (aumento ≥ 30 m), melhora no peptídeo natriurético pró-tipo B N-terminal (NT-proBNP) (diminuição no NT-proBNP ≥ 30% ou manutenção/obtenção do nível de NT-proBNP < 300 ng/L) e melhora da CF OMS ou manutenção da CF II da OMS. A progressão da doença foi medida pelo tempo até o óbito ou primeira ocorrência de um evento de piora clínica. Os eventos de piora clínica incluíram inclusão na lista para transplante pulmonar e/ou cardíaco, necessidade de iniciar terapia de resgate com uma terapia de HAP de base aprovada ou a necessidade de aumentar a dose de prostaciclina infusional em ≥ 10%, necessidade de septostomia atrial, hospitalização por piora da HAP (≥ 24 horas), ou deterioração da HAP (piora da CF da OMS e redução da distância do TC6M ≥ 15%, com ambos os eventos ocorrendo ao mesmo tempo ou em momentos diferentes). Os eventos de piora clínica e óbito foram registrados até que o último paciente completasse a visita da Semana 24 (dados até a linha de corte dos dados; duração mediana da exposição de 33,6 semanas).
Os pacientes tratados com Sotatercepte apresentaram melhora clínica estatisticamente significativa, melhora na CF da OMS e atraso na progressão da doença, incluindo redução do risco de morte e hospitalização em comparação aos pacientes tratados com placebo (veja Tabela 1, Tabela 2 e Figura 2).
Tabela 1: Resultados secundários de eficácia do estudo STELLAR
Desfecho
Placebo (N=160)
Sotatercepte (N=163)
IC 95%
Valor de p
Proporção de pacientes que obtiveram melhora multicomponente* (MMC) a partir da linha de base na Semana 24, n (%)
16 (10,1)
63 (38,9)†
N/A
< ,001‡
Alteração da RVP basal na Semana 24 (ASE) (dinas*seg/cm^5)
N/A
-234,6 (27,5)§
(-288,4; -180,8)
< ,001¶
Alteração dos níveis basais de NT-proBNP na Semana 24 (ASE) (pg/mL)
N/A
-441,6 (67,3)#
(-573,5; -309,6)
< ,001¶
Proporção de pacientes que melhoram a CF desde o início na Semana 24, n (%)
22 (13,8)
48 (29,4)†
N/A
< ,001‡
Tempo até o óbito ou a primeira ocorrência de um evento de piora clínicaÞ, n (%)
42 (26,3)
9 (5,5)
0,163 (0,076; 0,347)ß
< ,001à
Proporção de pacientes que mantiveram ou alcançaram um escoreè de baixo risco na Semana 24 versus linha de base, n (%)
29 (18,2)
64 (39,5)
N/A
< ,001‡
Mudança da linha de base na pontuação do domínio de impactos físicos da HAP SYMPACT®ð na Semana 24 (ASE)
N/A
-0,26 (0,115)ø
-(0,490; -0,040)
< ,010¶
Mudança da linha de base no escore do domínio de sintomas cardiopulmonares da HAP SYMPACT®ð na Semana 24 (ASE)
N/A
-0,13 (0,062)ø
(-0,256; -0,014)
0,028¶
Mudança em relação à linha de base no escore do domínio de impactos cognitivos/emocionais da HAP SYMPACT®ð na Semana 24 (ASE)
N/A
-0,16 (0,123)ø
(-0,399; 0,084)
0,156¶
ASE = erro padrão assintótico. Nota: Sempre que foram utilizados fatores de randomização estratificados, os fatores de randomização estratificados foram CF da OMS de base (Classe II ou III) e terapia de base para HAP (terapia mono/dupla ou tripla). *Um paciente satisfaz o MMC se todos os sintomas seguintes ocorrerem na Semana 24 em relação ao valor basal: melhora no TC6M (aumento ≥ 30 m), melhora no NT-proBNP (redução ≥ 30% ou manutenção/obtenção de NT-proBNP < 300 pg/mL) e melhora da CF da OMS ou manutenção da CF II da OMS. † Um resultado ausente na Semana 24, não devido à COVID-19, foi considerado um não respondedor. Os indivíduos que faltaram às avaliações devido à COVID-19 foram retirados do denominador. ‡ A comparação com placebo utiliza o método Cochran-Mantel-Haenszel (CMH) estratificado por fatores de randomização. § Estimativa de mudança de localização de Hodges-Lehmann em relação ao placebo (mediana de todas as diferenças pareadas). A mudança em relação ao valor inicial da RVP na Semana 24 para indivíduos que faleceram foi atribuída como 20.000 para receber a pior classificação. A mudança em relação ao valor inicial da RVP na Semana 24 para indivíduos que tinham dados faltantes devido a um evento de agravamento clínico não fatal foi imputada como 15.000 para receber a próxima pior classificação. ¶ O valor p de Wilcoxon refere-se ao valor p do teste de Wilcoxon estratificado por classificação alinhados com fatores de randomização como estratos. # Estimativa de mudança de localização de Hodges-Lehmann em relação ao placebo (mediana de todas as diferenças pareadas). A mudança da linha de base no NT-proBNP na Semana 24 para indivíduos que morreram foi atribuída como 200.000 para receber a pior classificação. A mudança em relação à linha de base no NT-proBNP na Semana 24 para indivíduos que tinham dados faltantes devido a um evento de agravamento clínico não fatal foi imputada como 150.000 para receber a próxima pior classificação. Þ Tempo até o óbito ou a primeira ocorrência de qualquer um dos seguintes eventos de piora clínica: a)piora relacionada à inclusão em lista para transplante de pulmão e/ou coração, b) necessidade de iniciar terapia de resgate com uma terapia HAP de base aprovada ou a necessidade de aumentar a dose de prostaciclina em 10% ou mais, c) necessidade de septostomia atrial, d) hospitalização por agravamento da HAP (≥ 24 horas), e) deterioração da HAP definida por ambos os seguintes eventos ocorridos a qualquer momento (mesmo que tenham começado em momentos diferentes) em comparação com seus valores basais: piora da CF da OMS e diminuição da distância no TC6M em ≥ 15%, confirmada por 2 exames com pelo menos 4 horas de intervalo, mas não mais de 1 semana. ß A razão de risco (Sotatercepte/placebo) foi derivada de um modelo de risco proporcional de Cox com grupo de tratamento como covariável estratificada pelos fatores de randomização. à Comparação do teste log-rank com placebo estratificado pelos fatores de randomização. è Utilização de calculadora do escore de risco francesa. ð Hipertensão Arterial Pulmonar – Sintomas e Impacto. ø Mudança de localização de Hodges-Lehmann em relação à estimativa do placebo (mediana de todas as diferenças pareadas). A mudança em relação à linha de base nas pontuações do SYMPACT na Semana 24 para indivíduos que morreram foi assinalada como 200 para receber a pior classificação. A mudança em relação à linha de base nos escores do SYMPACT na Semana 24 para indivíduos que tinham dados faltantes devido a um evento de agravamento clínico não fatal foi imputada como 150 para receber a próxima pior classificação.
Tabela 2: Morte ou Eventos de Piora Clínica
-
Placebo (N=160)
Sotatercepte (N=163)
Número total de indivíduos que sofreram óbito ou pelo menos um evento de piora clínica, n (%)
42 (26,3)
9 (5,5)
Avaliação de óbito ou primeira ocorrência de piora clínica*, n (%)
Morte
6 (3,8)
2 (1,2)
Piora relacionada à inclusão em lista para transplante de pulmão e/ou coração
1 (0,6)
1 (0,6)
Necessidade de iniciar terapia de resgate com terapia de HAP aprovada ou necessidade de aumentar a dose de prostaciclina por infusão em 10% ou mais
17 (10,6)
2 (1,2)
Necessidade de septostomia atrial
0 (0,0)
0 (0,0)
Hospitalização específica para HAP (≥ 24 horas)
7 (4,4)
0 (0,0)
Deterioração da HAP†
15 (9,4)
4 (2,5)
* Um indivíduo pode ter mais de uma avaliação registrada para seu primeiro evento de piora clínica. Houve 3 indivíduos que receberam placebo e 0 indivíduo que recebeu sotatercepte que tiveram mais de uma avaliação registrada para seu primeiro evento de piora clínica. † A deterioração da terapia para HAP é definida por ambos os seguintes eventos ocorrendo a qualquer momento, mesmo que tenham começado em momentos diferentes, em comparação com seus valores basais: (a) Piora da classe funcional da OMS (II para III, III para IV, II para IV, etc.); e (b) Diminuição da distância no TC6M em ≥ 15% (confirmada por dois TC6M com pelo menos 4 horas de intervalo, mas não mais do que uma semana). N = número de indivíduos da população FAS; n = número de indivíduos da categoria. As porcentagens são calculadas como (n/N)*100.
Figura 2: Tempo até a morte ou primeira ocorrência de eventos de piora clínica - Gráfico de Kaplan-Meier
Referências Bibliográficas
Hoeper MM, Badesch DB, Ghofrani HA, et al. Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension. The New England Journal of Medicine 2023.
Características Farmacológicas
Classe terapêutica: Sotatercepte (sotatercepte) é um inibidor de sinalização de activina.
Mecanismo de Ação
O sotatercepte é um inibidor da sinalização de activina com alta seletividade para activina A, uma glicoproteína dimérica que pertence à superfamília de ligantes do fator de crescimento transformador β (TGF-β). A activina A se liga ao receptor de activina tipo IIA (ActRIIA) regulando a sinalização chave para inflamação, proliferação celular, apoptose e homeostase tecidual.
Os níveis de activina A estão aumentados em pacientes com HAP. A ligação da activina ao ActRIIA promove sinalização proliferativa enquanto há uma diminuição na sinalização do receptor morfogenético ósseo tipo II (BMPR-II) antiproliferativo. O desequilíbrio da sinalização ActRIIA-BMPRII subjacente à HAP resulta em hiperproliferação de células vasculares, causando remodelamento patológico da parede arterial pulmonar, estreitamento da luz arterial, aumento da resistência vascular pulmonar, aumento da pressão da artéria pulmonar e disfunção ventricular direita.
O sotatercepte consiste em uma proteína de fusão do receptor homodimérico de activina recombinante tipo IIA-Fc (ActRIIA-Fc) que atua como um captador do ligante que elimina o excesso de activina A e outros ligantes para ActRIIA para inibir a sinalização de activina. Como resultado, o sotatercepte reequilibra a sinalização pró-proliferativa (ActRIIA/Smad2/3-mediado) e antiproliferativa (BMPRII/Smad1/5/8 mediado) para modular a proliferação vascular. Em modelos de HAP em ratos, um análogo do sotatercepte reduziu a expressão de marcadores pró-inflamatórios na parede arterial pulmonar, reduziu o recrutamento leucocitário, inibiu a proliferação de células endoteliais e musculares lisas e promoveu apoptose na vasculatura doente. Essas alterações celulares foram associadas a paredes mais finas dos vasos, remodelamento arterial e ventricular direito reversos e melhora hemodinâmica. Em estudos clínicos de HAP, Sotatercepte diminuiu a resistência vascular pulmonar e reverteu o remodelamento do ventrículo direito.
Farmacodinâmica
Um estudo clínico de fase 2 avaliou a resistência vascular pulmonar (RVP) em pacientes com HAP após 24 semanas de tratamento com sotatercepte. A diminuição em relação ao valor basal na RVP foi significativamente maior nos grupos sotatercepte 0,7 mg/kg e 0,3 mg/kg em comparação com o grupo placebo. A diferença média dos mínimos quadrados (MQ) ajustados por placebo em relação ao valor basal foi de -269,4 dinas*seg/cm5 (IC 95%: -365,8; -173,0) para o grupo sotatercepte 0,7 mg/kg e -151,1 dinas*seg/cm5 (IC 95%: -249,6; -52,6) para o grupo sotatercepte 0,3 mg/kg. No STELLAR, a diminuição em relação ao valor basal da RVP foi também significativamente maior no grupo sotatercepte 0,7 mg/kg em comparação com o grupo placebo.
Farmacocinética
Em pacientes com HAP, a média geométrica (%CV) da área sob a curva (AUC) em estado estável e a concentração de pico em estado estável (Cmáx) na dose de 0,7 mg/kg a cada 3 semanas (Q3W) foram 171,3 mcg×d/mL (34,2%) e 9,7 mcg/mL (30%CV), respectivamente. A AUC e Cmáx do sotatercepte aumentam proporcionalmente com a dose. O estado de equilíbrio é alcançado após aproximadamente 15 semanas com a dosagem múltipla Q3W. A razão de acumulação da AUC do sotatercepte foi de aproximadamente 2,2.
Absorção
A formulação SC tem uma biodisponibilidade absoluta de aproximadamente 66%. A concentração máxima de sotatercepte é atingida em um tempo mediano até o pico da concentração do medicamento (Tmáx) de aproximadamente 7 dias (intervalo de 2 a 8 dias) após doses SC múltiplas (0,1 mg/kg a cada 4 semanas) em mulheres na pós-menopausa.
Distribuição
O volume central de distribuição (%CV) do sotatercepte é de aproximadamente 3,6 L (24,7%). O volume periférico de distribuição (%CV) é de aproximadamente 1,7 L (73,3%).
Eliminação
A depuração do sotatercepte é de aproximadamente 0,18 L/dia. A média geométrica da meia-vida terminal (%CV) é de aproximadamente 21 dias (33,8%).
Metabolismo
O sotatercepte é catabolizado por processos gerais de degradação de proteínas.
Populações Especiais
Idade, sexo e raça
Não foram observadas diferenças clinicamente significativas na farmacocinética (PK) do sotatercepte com base na idade (18 a 81 anos de idade), sexo ou raça.
Peso Corporal
A depuração (CL) e o volume central de distribuição (Vc) do sotatercepte aumentaram com o aumento do peso corporal. O regime posológico baseado no peso recomendado resulta em exposições consistentes ao sotatercepte, independentemente do peso corporal.
Insuficiência Renal
A PK do sotatercepte foi comparável em pacientes com HAP com insuficiência renal leve a moderada (TFGe variando entre 30 e 89 mL/min/1,73m2) e naqueles com função renal normal (TFGe ≥ 90 mL/min/1,73 m2). Além disso, a PK do sotatercepte é comparável entre pacientes com doença renal terminal (DRCT) sem HAP e pacientes com função renal normal. Sotatercepte não é dialisável durante a hemodiálise. Não é recomendado qualquer ajuste posológico em pacientes com insuficiência renal. O sotatercepte não foi estudado em pacientes com HAP com insuficiência renal grave (TFGe < 30 mL/min/1,73 m2).
Insuficiência hepática
Não é esperado que a insuficiência hepática (determinada pela Classificação de Child-Pugh) influencie o metabolismo do sotatercepte, uma vez que o sotatercepte é metabolizado através do catabolismo celular. O sotatercepte não foi estudado em pacientes com HAP com insuficiência hepática (Classificação de Child-Pugh A a C).
Imunogenicidade
A incidência observada de anticorpos antidrogas é altamente dependente da sensibilidade e especificidade do ensaio. Diferenças nos métodos de ensaio impedem comparações significativas da incidência de anticorpos antidrogas no estudo descrito abaixo com a incidência de anticorpos antidrogas em outros estudos, incluindo os de Sotatercepte ou de outros produtos de sotatercepte.
Durante o período de tratamento de 24 semanas no estudo pivotal (STELLAR), 44/163 (27%) dos pacientes tratados com sotatercepte desenvolveram anticorpos anti-sotatercepte. Entre esses 44 pacientes, 12 (27%) testaram positivo para anticorpos neutralizantes contra o sotatercepte. Os anticorpos anti-sotatercepte geralmente tinham títulos baixos, com um título mediano de 30 (variação < 20 a 640).
Não foram identificados efeitos clínicos dos anticorpos anti-sotatercepte na farmacocinética, farmacodinâmica, segurança ou efetividade do sotatercepte durante o período de tratamento de 24 semanas.
Toxicologia animal
Toxicidade aguda
Nenhuma toxicidade aguda foi observada em estudos de toxicidade SC de dose repetida em dosagens de até 30 mg/kg em ratos e 50 mg/kg em macacos (doses únicas proporcionaram exposições aproximadamente 15 vezes e 38 vezes, respetivamente, à exposição humana na dose humana máxima recomendada (DHMR) (com base na AUC estimada)).
Toxicidade crônica
Em ratos e macacos, os estudos de toxicidade SC de maior duração foram de 3 meses e 9 meses, respectivamente. Em ratos administrados com doses semanais de 0,3, 3 e 30 mg/kg durante 3 meses, os achados adversos incluíram degeneração do ducto eferente/testicular, congestão/necrose da glândula adrenal, glomerulonefrite membranoproliferativa e nefrite tubulointersticial nos rins que ocorreram com uma exposição 18 vezes maior que a DHMR (com base na AUC estimada). Tanto as alterações adrenais quanto as renais demonstraram reversibilidade após um período de recuperação de 1 mês. Em macacos administrados com 1, 2,6 e 10 mg/kg uma vez a cada 4 semanas e 10 mg/kg uma vez a cada 2 semanas, as alterações adversas foram limitadas à glomerulonefrite e nefrite tubulointersticial nos rins que ocorreram em exposições ≥ 6 vezes a DHMR (com base na ASC estimada). As alterações renais em macacos se resolveram parcialmente após um período de recuperação de 3 meses.
Carcinogênese
Não foram realizados estudos de carcinogenicidade com o sotatercepte.
Mutagênese
Não foram realizados estudos de mutagenicidade com o sotatercepte.
Reprodução
Em um estudo de fertilidade e desenvolvimento embrionário inicial em ratas, o sotatercepte foi administrado SC uma vez por semana nas doses de 5, 15 e 50 mg/kg a partir de 2 semanas antes do acasalamento e até ao dia 7 de gestação. Em doses ≥ 15 mg/kg (≥ 9 vezes o DHMR, com base na AUC estimada), as taxas de gravidez foram reduzidas e houve aumentos na perda pré e pós-implantação e reduções no tamanho da ninhada viva. O aumento da duração do ciclo estral ocorreu apenas com 50 mg/kg (21 vezes o DHMR, com base na AUC estimada).
Em um estudo de fertilidade em ratos machos, o sotatercepte foi administrado SC uma vez por semana nas doses de 0,3, 3 e 30 mg/kg durante 13 semanas (começando 10 semanas antes do acasalamento). Um subgrupo de animais foi examinado após um período de recuperação de 13 semanas. Com ≥ 0,3 mg/kg (0,5 vezes o DHMR, com base na AUC estimada) houve alterações histológicas irreversíveis nos ductos eferentes, testículos e epidídimos. Ocorreram diminuições reversíveis na fertilidade a 30 mg/kg (20 vezes o DHMR, com base na AUC estimada).
Desenvolvimento
Em estudos de toxicidade para o desenvolvimento embriofetal, animais gestantes foram dosados por via subcutânea com sotatercepte durante o período de organogênese. O sotatercepte foi administrado a ratas nos dias 6 e 13 de gestação nas doses de 5, 15 ou 50 mg/kg e a coelhos nos dias 7e 14 de gestação nas doses de 0,5, 1,5 ou 5 mg/kg. Os efeitos em ambas as espécies incluíram reduções no número de fetos vivos e no peso corporal fetal, atrasos na ossificação e aumentos nas reabsorções e perdas pós-implantação. Em ratos e coelhos, estes efeitos foram observados em exposições (com base na área sob a curva (AUC)) aproximadamente 4 vezes e 0,6 vezes a dose humana máxima recomendada (DHMR), respetivamente. Apenas em ratos, as variações esqueléticas (aumento do número de costelas supranumerárias e mudanças no número de vértebras torácicas ou lombares) ocorreram em uma exposição 15 vezes maior do que a exposição humana no DHMR.
Em um estudo de desenvolvimento pré e pós-natal em ratos, o sotatercepte foi administrado por via subcutânea nas doses de 1,5 e 5 mg/kg nos dias 6 e 13 de gestação, ou nas doses de 1,5, 5 ou 10 mg/kg durante a lactação nos dias 1, 8 e 15. Não houve efeitos adversos em filhotes de primeira geração filial (F1) de mães dosadas durante a gestação em exposições estimadas até 2 vezes a DHMR. Em filhotes F1 de mães dosadas durante a lactação, as diminuições no peso dos filhotes correlacionaram-se com atrasos na maturação sexual em exposições estimadas (com base na AUC) ≥ 2 vezes o DHMR.
Como devo armazenar o Winrevair?
Armazenar em geladeira (2°C e 8°C). Não congelar. Manter nesta embalagem até o final do uso. Manter na embalagem original para proteger da luz.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Contate o seu médico ou farmacêutico se o kit de injeção não utilizado esteve fora da geladeira durante um longo período de tempo.
Após você misturar o pó do medicamento com a água para injetáveis (fornecida no kit), é recomendável que você o injete imediatamente, mas em até 4 horas após a reconstituição.
Após preparo, utilize a solução reconstituída o mais rápido possível, mas em até 4 horas após a reconstituição, em temperatura de até 30°C.
A estabilidade por até 4 horas foi comprovada para os aspectos físico-químicos do medicamento. Do ponto de vista microbiológico, recomenda-se que o medicamento seja utilizado imediatamente após a reconstituição.
Características do medicamento
WinrevairTM é um pó liofilizado estéril, sem conservantes, branco a esbranquiçado, para administração subcutânea após reconstituição. Após a reconstituição, WinrevairTM é límpido a opalescente, incolor a ligeiramente amarelado acastanhado e livre de grumos ou pó.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Mensagens de Alerta do Winrevair
Por favor, leia esta informação cuidadosamente antes de começar a usar o seu medicamento, mesmo que você já esteja utilizando o medicamento. Algumas das informações podem ter sido alteradas.
Dizeres Legais do Winrevair
Registro 1.0171.0238
Importado e Registrado por: Merck Sharp & Dohme Farmacêutica Ltda. Av. Dr. Chucri Zaidan, 296 – São Paulo/SP CNPJ 03.560.974/0001-18 - Brasil
O conteúdo desta bula foi extraído manualmente da bula original, sob supervisão técnica do(a) farmacêutica responsável: Karime Halmenschlager Sleiman (CRF- PR 39421). Última atualização: 18 de Março de 2025.